作業メモ
ここからは勉強過程で残した作業用メモです。参考になるかもしれないし、ならないかもしれません。
GPUあるいはマルチスレッドの検証
GPGPUが本当にどれぐらい効くのか、またマルチスレッドはどのぐらい効くのか、それを検証してみます。
実行可能ファイルのファイル名の変更方法
- 結論:よくわからなかった。cmakeのコマンドをいじれば実行可能ファイル名を変えられるはずなんだけど、CMakeLists.txtの中にもそれらしきものは見つからず…
- LAMMPS_BINARYやOUTPUT_NAMEに実行可能ファイル名が格納されているかもしれない。
- ここに書いてあった。
-D LAMMPS_MACHINE=nameのビルドオプションを付けるとsuffixがつくらしい。(lmp_nameみたいに)
KOKKOSインストール手順
基本はここに書いてあるので参照するべし。
- 利用可能なアーキテクチャ一覧がある(こちらから参照)
- 今回の場合はRyzen 5950XとNVIDIA GeForce GTX 1650 or RTX 3090なのでArch-IDの中でも
ZEN3とTURING75かAMPERE86が利用可能CCはCompute Capabilityの略でここから参照可能AMPERE80は過去の遺物?
cmakeのビルド時に以下のコードの入力する必要がある- 連続して入力する場合には
cmake -D PKG_KOKKOS=yes -D Kokkos_ARCH_ZEN3=yes ../cmakeのようにする - 共通
-D PKG_KOKKOS=yes - Zen3 CPU向け
-D Kokkos_ARCH_HOSTARCH=yes # HOSTARCHはここではZEN3 -D Kokkos_ENABLE_OPENMP=yes -D BUILD_OMP=yes - NVIDIA CPU向け
-D Kokkos_ARCH_GPUARCH=yes # GPUARCHはここではTURING75かAMPERE86 -D Kokkos_ENABLE_CUDA=yes -D CMAKE_CXX_COMPILER=${HOME}/lammps/lib/kokkos/bin/nvcc_wrapper
- 連続して入力する場合には
- 今回の場合はRyzen 5950XとNVIDIA GeForce GTX 1650 or RTX 3090なのでArch-IDの中でも
- コンパイルする
make -j 4- なんかいろいろ警告が出るけど大きく分けてこの2つ
nvcc warning : The 'compute_35', ~~~: nvccのコンパイラに-Xcompiler "/wd 4819"を渡せば解決するらしいがcmakeからどのように設定するのか不明- 多分CMakeLists.txtを書き換えればいいのかな…
warning #1675-D: unrecognized GCC pragma: これもnvccのコンパイラに-Xcudafe "--diag_suppress=unrecognized_gcc_pragma"を通せばいいみたい
- なんかいろいろ警告が出るけど大きく分けてこの2つ
mpirun -np 16とGPUを変更した際の実行結果
結果の前に実行した際の疑問点を以下に…
- OpenMPのスレッドはどのようにして増やす??
- GPUが使われているようには思えないんだけど…
mpirun -np 16 $LAMMPS_BUILD_DIR/lmp -sf gpu -pk gpu 1 -in in.pour.flatwallの-sf gpu -pk gpu 1が肝心- ただメモリ使用量が増えるだけでCUDAは使われないようだ…
- granularモジュールはCUDAを使えない??
- 正解。各
pair_styleのコマンドの中に使用できるAcceleratorが書かれている(たとえばgranのページ)granularコマンドはいずれのAcceleratorも使えないgran/hookeとpair_style gran/hertz/historyコマンドはomp(OpenMP)のみgran/hooke/historyコマンドはompとkk(KOKKOS)のみ- でもそれぞれのコマンドの解説ページには以下のように書かれてる…
-
Styles with a gpu, intel, kk, omp, or opt suffix are functionally the same as the corresponding style without the suffix.
-
- じゃあKOKKOSってなによ…
- じゃあ
gran/hooke/historyとそれ以外のコマンドって何が違うのよ…historyはせん断応力が履歴をもたらすという意味らしい- $F_{hk}=\left(k_n\delta\boldsymbol{n}{ij}-m{eff}\gamma_n\boldsymbol{v}n\right) - \left(k_t\Delta\boldsymbol{s}_t+m{eff}\gamma_t\boldsymbol{v}_t\right)$のうちの$k_t\Delta\boldsymbol{s}_t$の部分
- ここで$\Delta\boldsymbol{s}_t$は2粒子間の接線変位ベクトル(降伏関数を満たすように切り捨てたもの←ここ重要!)
- 粘性によるエネルギー損失だけでなく塑性変形によるエネルギー損失が考慮できる!
- しかし計算コストのかかりそうな
gran/hooke/historyのみがKOKKOSに対応しているのはなぜだ…
- $F_{hk}=\left(k_n\delta\boldsymbol{n}{ij}-m{eff}\gamma_n\boldsymbol{v}n\right) - \left(k_t\Delta\boldsymbol{s}_t+m{eff}\gamma_t\boldsymbol{v}_t\right)$のうちの$k_t\Delta\boldsymbol{s}_t$の部分
- そもそも
granularとgranの違いって何よ…- (まだ調べてない…)
mpirun -np 32と設定するとエラーが出るんだけどなんで??mpirun -np 16が限界だったから物理コアに依存?- ここを見ると
processing elementは何もオプションを指定しない場合はプロセッサーのコアになるらしい。-
By default, Open MPI defines that a “processing element” is a processor core. However, if –use-hwthread-cpus is specified on the mpirun command line, then a “processing element” is a hardware thread.
-
研究室内の自分PCにwslをインストール使用としたがうまくいかない…
ファイル システムの 1 つをマウント中にエラーが発生しました。詳細については、「dmesg」を実行してください。- ここを見たけど
wsl --updateでも改善しない… dmesg | grep -i error(dmesgはLinuxカーネルが起動時に出力したメッセージを表示するコマンド)をやってみた[ 2.678154] EXT4-fs (sdc): mounted filesystem with ordered data mode. Opts: discard,errors=remount-ro,data=ordered [ 3.307626] init: (1) ERROR: MountPlan9WithRetry:285: mount drvfs on /mnt/f (cache=mmap,noatime,msize=262144,trans=virtio,aname=drvfs;path=F:\;uid=0;gid=0;symlinkroot=/mnt/ [ 3.967180] init: (1) ERROR: MountPlan9WithRetry:285: mount drvfs on /mnt/f (cache=mmap,noatime,msize=262144,trans=virtio,aname=drvfs;path=F:\;uid=0;gid=0;symlinkroot=/mnt/- なんか
path=F:\っておかしくね… - Fドライブはフォーマットされていない8TBのHDDドライブ
- 多分以前何かを入れておいて初期化したままになっていたみたい…
- フォーマットしてバックアップドライブとして指定したら解決!(多分空き容量か何かで見てる??)
- ここを見たけど
bin2cエラー
CMake Error at Modules/Packages/GPU.cmake:44 (message):
Could not find bin2c, use -DBIN2C=/path/to/bin2c to help cmake finding it.
このエラーが無くならない
- 再インストールをしてもだめ
- bin2cへのパスを記入せよと言われているが、そのbin2cのファイルが見当たらない
- everythingで探していたからだった…
- everythingのオプション→検索データ→フォルダ→すべてをすぐに更新
- データベースが更新され
\\wsl.localhost\Ubuntu\usr\local\cuda-11.7\bin\bin2cにあることが分かった
- everythingで探していたからだった…
別PC(5950XとRTX3070)でセットアップした際のエラー
make -j 16コンパイル中に以下のようなエラーcc1plus: error: bad value (‘znver3’) for ‘-march=’ switch cc1plus: note: valid arguments to ‘-march=’ switch are: nocona core2 nehalem corei7 westmere sandybridge corei7-avx ivybridge core-avx-i haswell core-avx2 broadwell skylake skylake-avx512 cannonlake icelake-client icelake-server cascadelake tigerlake bonnell atom silvermont slm goldmont goldmont-plus tremont knl knm x86-64 eden-x2 nano nano-1000 nano-2000 nano-3000 nano-x2 eden-x4 nano-x4 k8 k8-sse3 opteron opteron-sse3 athlon64 athlon64-sse3 athlon-fx amdfam10 barcelona bdver1 bdver2 bdver3 bdver4 znver1 znver2 btver1 btver2 native; did you mean ‘znver1’?- gccのバージョンが低いことが理由
- 下記のコードで解決できるらしいですが、理由までは調べ切れていません。
sudo apt install gcc-10 g++-10 sudo update-alternatives --install /usr/bin/gcc gcc /usr/bin/gcc-10 100 --slave /usr/bin/g++ g++ /usr/bin/g++-10 --slave /usr/bin/gcov gcov /usr/bin/gcov-10 sudo update-alternatives --config gcc
- 下記のコードで解決できるらしいですが、理由までは調べ切れていません。
Ghost Atomについて
- このページに書いてあった。
computeとthermoの違いが分からない…
- どのように変数を計算させるのか、どのようにそれらをログとして出力するのか
- 3種類の物理量:
global、per-atom、localがあるglobal:thermo_style customで出力可能per-atom:dump customで出力可能local:compute reduceで出力可能
- では杭壁面に作用する応力はどの変数に当たるのか?
- たとえばcompute pressureというコマンドがある
- ここで計算される圧力あるいは応力テンソルは次のような形をしている
- $P=\frac{Nk_BT}{V}+\frac{\sum_i^{N’}r_i \cdot f_i}{dV}$
- 右辺第1項は状態方程式による項、第2項はビリアル定理による項
- ビリアル定理は「系全体の運動エネルギー$K$の時間平均は、系全体のポテンシャルエネルギー$V$の時間平均の$1/2$に等しい」ことを示す
- $P=\frac{Nk_BT}{V}+\frac{\sum_i^{N’}r_i \cdot f_i}{dV}$
- 何か計算したいものと違う気がする…
- 粒状体では温度は考慮しないから、第2項しか存在しないはず
- 各粒子に作用する力は重力と隣接する粒子からの接触力に起因するもの(?)
- 多分このコマンドは複数の領域に分割されるようなケースは想定していないのでは…?
- 簡単なコードで解析してみたい
- ここで計算される圧力あるいは応力テンソルは次のような形をしている
- また
reduceというコマンドがある- localあるいはper-atomの出力された物理量をまとめられる
- これ(compute temp/sphere)かもしれない -
- たとえばcompute pressureというコマンドがある
- 3種類の物理量:
- おそらくthermoから見た方がいいかもしれない
別のマニュアル(Rawiwan, 2021)から得られたこと
- 以下のソフトをインストール
- VMware Workstation
- Ubuntu desktop
- Paraview v5.9: 粒子の動きを描画するソフト
- Matlab: 応力状態や間隙比などを計算するためのプログラム
bash LAMMPS_Build_Script_Serial.shが実行されている。- 詳しいことは読み取れていないが、やっていることはgranularモジュールを含んだ形でのシリアル(直列)処理とMPI用実行可能ファイルの作成?
-
rm -f lmp_serial # cd src/STUBS rm -f libmpi_stubs.a rm -f mpi.o make -s cd .. make yes-GRANULAR echo 'Compiling mpi...' make serial >&Lammps_Output_File echo 'Cleaning up files...' %make clean-all mv lmp_serial .. echo 'Done!' cd .. echo `date` >>Build_Dates.txt sleep 2
- 解析するのに用いているファイルは
.inファイル、.ljファイル、実行可能ファイル(lmp_mpiかlmp_serial).inファイルには載荷条件が書かれている- デフォルトにない設定項目が
in.isotropic内には存在する→恐らく新たに追加されたコマンドpair_modify trace_energy: 接触点ごとのエネルギー項をシミュレーション中に出力するコマンドらしい- 一種のフラグとして機能している(
if (trac_energy)のように) - このフラグによって計算されるかどうかが左右されるのは
shearという変数gitLAMMPS_2019_Rigid_mpi_Ver8だとshear[3]からshear[8]、shear[26]からshear[29]が更新されている- この
shearが定義されているコードは発見できなかった
- なぜパラメータ計算に関するコマンドが
pair_modifyで定義されているのか?computeじゃないのか?
- 一種のフラグとして機能している(
- 他にも
run_style verletやtimestep auto、fix ID GROUP-ID multistressなどがある fix ID GROUP-ID multistress: 複数の応力条件や等体積条件を満たすように境界領域をコントロールする。-
fix ID group-ID multistress N M R keyword ID, group-ID are documented in "fix"_fix.html command multistress = style name of this fix command N = the percentage error in the target stress. See note 1. M = the number of steps to run within N% of the target stress before displaying that the target stress has been attained. See note 1. R = optional but highly recommended - this is the maximum engineering strain rate permissible for stress-controlled boundaries. one or more keyword/value pairs must be appended
-
- ここに境界領域の応力の求め方が書いてあるはず…(詳しく見たいので別項で)
- デフォルトにない設定項目が
.ljファイルには粒子の初期座標が書かれている- 生のテキストファイルとして読めるけどファイル構造は結構簡単そう
- ヘッダーに粒子の種類数、粒子数、存在範囲
- そのあとに各々の粒子の情報(種類ID、分からん、分からん、x, y ,z)
- ここに書いてあった。atom typeがsphereの場合は次の通り→
atom-ID atom-type diameter density x y z
- 生のテキストファイルとして読めるけどファイル構造は結構簡単そう
fix ID GROUP-ID multistressをもう少し詳しく見る
- 全体的な流れとしては6成分の応力を計算した後、if文で応力増分が一定の値に落ち着くまでひずみを最急降下法とかで最適化しているはず
- 等体積条件での制御も同じ
- 参照したファイルは
fix_multistress.cpp constantpq_loop?は三項演算子theratesというのに上限と下限を設けているfirstidとsecondidが何か
eval_fix_deform_params_constantpqでconstantpq_loopは使用されている。theratesはtemprates: テンプレートではなくtemporal strain rateの略tempratesの計算方法は二つ- 目標応力と現在応力の差分に体積弾性率を掛けたもの
- なんでパラメータ名が
tallymeansなの?何の略? meaneffectivestress = (tallymeans[0] + tallymeans[1] + tallymeans[2]) / 3.0という書き方を見ると応力テンソルの対角成分のことを意味するらしい- なぜdiagonal componentとかではないのか…?
- headerファイルに
The total mean stresses from all processorsと記載されていた fix_multistress.cpp内にはtallymeansは計算されていない
- 1次元配列で要素数は6個、上三角行列の各成分に対応 - どこで計算されているんだ…?
- なんでパラメータ名が
multistress以降のパラメータとしてmaxrate
- 目標応力と現在応力の差分に体積弾性率を掛けたもの
MPI_Allreduce(&means[0], &tallymeans[0], 6, MPI_DOUBLE, MPI_SUM, world);この式はどういう意味?MPI_ALLREDUCとはMPI_REDUCEの結果を全プロセスに渡すという意味MPI_REDUCEとはMPI_GATHERと演算を組み合わせたもの.例えば,各々のプロセスのデータの和,積,最大値,最小値などを求め,1つのプロセスに渡す.- そもそもMPIとは…
- Message Passing Interfaceの略。
- スレッドとメモリをひとまとめにしたものをプロセスと呼ぶ
- MPIはプロセス間のメモリ情報のやり取りを規定したもの
MPI_GATHERとは各々のプロセスのデータを連結して1つのプロセスに集める- 引数は次の通り→
MPI_Allreduce(sendbuf, recvbuf, count, datatype, op, comm, ierror)- countとはデータの個数のこと
- commはどのプロセス間での通信を許可するかの範囲を決めたもの(理解が曖昧)。デフォルトで
MPI_COMM_WORLDが用意されている。今回のworldもその派生?
- (なるほど!)
sendbufとrecvbufはそれぞれ送信するデータと受信するデータの先頭のアドレスを引数とする- だから
means[i]みたいな書き方にはならない - ちなみに
meansは「現在のタイムステップにおける平均応力」
- だから
- なのでこのコードでやっていることを日本語で記述すると「各プロセス内の
meansに関して総和を取ったものを各プロセス内のtallymeansにブロードキャスト(同じ値を代入)する」
meansはどうやって計算されている?fix_mutilstress.hだとdouble *means; //~ The mean stresses on the current timestepと書かれている- ヘッダーファイルの変数に付くアスタリスクってどういう意味だっけ?
- ポインタだ
- 例えばこんな書き方になっている→
double *means = stressatom->array_export(); - アロー演算子(
->)はポインタからでも構造体や共用体の要素にアクセスできる。 - (特定の粒子あるいは粒子全体に作用する応力の総和によって、領域に作用する圧力を計算しているとすれば、杭に作用する応力ってどうやって計算するんだ…?)
stressatomはcomputeクラスでcompute_stress_atom.cppとcompute_stress_atom.hに定義されている- 詳しい計算は
array_export()メンバ内に書かれている
array_export()内の計算内容- (これかもしれない…)
if (mask[i] & groupbit){...}という構文がある...では各粒子の体積を次元を考慮して計算した後、stressと体積を掛け算して、stressの一次元目に対して総和を取っているstress[i][j]のうちiは粒子総数、jは応力テンソルの6成分
stress[i][j]はvatom[i][j]の総和…?- i番目の粒子に作用する力jによって生じる6成分の応力成分だったとたら
vatom[i][j][6]じゃないとおかしくない…?
- i番目の粒子に作用する力jによって生じる6成分の応力成分だったとたら
vatom[i][j]の定義はどうなっている- vはおそらくvirial(ビリアル)の略…ビリアル応力
- とりあえずlammpsの公式サイトを読
- あるいはこちらのサイトも参考になるかもしれない
- こちらに勉強内容を書いた
- 完全には理解できていないが、「ビリアル定理によって熱平衡状態にある多粒子系の応力テンソルは粒子間相互作用と粒子それぞれの運動エネルギーから定義できる」ことを指す
vatom[i][j]のうち[i]は粒子のインデックス、[j]は応力テンソルの6成分。(おそらくドキュメントに書かれている順番からxx, yy, zz, xy, xz, yzのはず)vatom[i][j]は粒子間相互作用以外にも結合力(bond)や結合角(angle)、二面角(dihedral), improper, kspaceからも計算される
compute_stress_atom.cpp内のif (mask[i] & groupbit){...}のmask[i]とgroupbitとはそれぞれなに?mask[i]はnlocal個の配列長さを持つnlocalはatom.h内にプロセッサーが有する粒子というコメントがある- lammpsでは解析する空間を区切っているがそれに関する名前が複数あって違いが良く分からない
- Region, Processor, Rank, Mask, Group, Neiborlist
mask[i] = (int) ubuf(buf[m++]).i;というコードがよく見かけられるubufは共用体として定義されていて、型変換を行っている- コンパイラによる警告を避けるためと書かれているけど、あまりよくない書き方なのでは…?あるいはbit数の異なるOSそれぞれに対応するための折衷案?
- 半分ぐらいは
unpack_borderの中に書かれている- 引数が
n, first, *buf→bufはポインタ渡し
- 引数が
m++はインクリメントなので、bufの中身が重要
- 条件付きの再帰関数になっている
- 再帰関数ではなくコールバック関数?
-
if (atom->nextra_border) for (int iextra = 0; iextra < atom->nextra_border; iextra++) m += modify->fix[atom->extra_border[iextra]]-> unpack_border(n,first,&buf[m]); - このコードが良く分からない…
nextra_boarderはおそらくnumber of particle to be extracted around borderの略bufは文字通り仮の変数で色々な者が入ってくる- 例えば粒子の位置であったり、速度であったり
pack_borderは粒子情報をバッファに保存し、近傍で再構築する際に通信するコマンドunpack_borderバッファから粒子情報を取り出すmask[i] = (int) ubuf(buf[m++]).i;というコードはバッファからのやり取りで本質的なものではない
mask[i]で境界領域の粒子の判別をしている?ではそのフラグはどのようにして切り替わっている?- 調べても情報は出てこなかった
fix_pour.cpp内ではatom->mask[n] = 1 | groupbit;と書かれている。|はbitでのOR演算子groupbitはcompute_stress_atom.cpp内でint groupbit = bitmask[igroup];と書かれているbitmaskはgroup.cpp内でfor (int i = 0; i < MAX_GROUP; i++) bitmask[i] = 1 << i;と書かれている- MAX_GROUPは32と定義されている
- これだとこのコードの後は’[11111111111111111111111111111111]’となっているはず
bitmaskの値を変更するようなコードは見つからない…
bitmaskは最大32個のグループを識別するもの- ということは
mask[i]もint型でないといけない
- ということは
- ここで打ち止め、もう少しlammpsのコードを触ってからこちらに戻ってくる。
fix wall_granコマンド- このリンクから
- ただKOKKOSでは対応していないコマンドのはずなのに、解析時にエラーがでないのはかなり変…
- (これかもしれない…)
lmp: error while loading shared libraries: libcuda.so.1の解決策について
- このエラーに行きつく前のエラー達
cmake -D CMAKE_CXX_COMPILER=${HOME}/lammps-stable_23Jun2022/lib/kokkos/bin/nvcc_wrapper ../cmakeのコマンドだけは別に実行しないと行けないらしい…- 理由は不明。とりあえずこのひとつ前のコードと分けて実行すると
Enable Packageのリストが正しく更新されることを確認済み。make -j 4の最初に表示されるビルド情報をしっかりと見ること
- 理由は不明。とりあえずこのひとつ前のコードと分けて実行すると
- そもそも
make installをすると新しくビルドをしてもう一度make installをしても更新されない現象がある- 解決策については以下に記載予定(現在編集中)
lammps配下のbuildフォルダを一旦削除~/.local/bin内にあるlmpを削除~/.profileのPATH=$PATH:$HOME/.local/binの行も一旦削除
- 現在調査中
- 解決策については以下に記載予定(現在編集中)
libcuda.so.1からlibcuda.soへのシンボリックリンクが作成されていないことが原因- ソースコードを修正するとしたら、ビルドの過程でシンボリックリンクを作成する必要があるがどうやるの?
libcuda.so自体はusr\local\cuda-11.7\targets\x86_64-linux\lib\stubs\libcuda.soに存在するが、libcuda.so.1は存在しない- ただ
libcuda.so.1がどの環境変数によって参照されているのかが分からない ubuntu20.04_gpuでは次のような記述がある-
# add missing symlink ln -s /usr/local/cuda-${CUDA_PKG_VERSION}/lib64/stubs/libcuda.so /usr/local/cuda-${CUDA_PKG_VERSION}/lib64/stubs/libcuda.so.1 libcuda.so.1からlibcuda.soへのシンボリックリンクが作成するコマンド- ただ
/usr/local/cuda-${CUDA_PKG_VERSION}/lib64/stubs/内にはlibcuda.so.1は存在していない- cudaが複数あるけどこれはよいのか…(
cuda/ cuda-11/ cuda-11.7/)
- cudaが複数あるけどこれはよいのか…(
- ちなみに
/usr/local/cuda-11.7$ ls -n lrwxrwxrwx 1 0 0 24 Jun 9 10:10 lib64 -> targets/x86_64-linux/libとなっているので
targets/x86_64-linux/lib配下のファイルはすべてlib64配下からシンボリックリンクとして作成されている
- ただ
-
- ひとまず
sudo ln -s libcuda.so libcuda.so.1コマンドをusr\local\cuda-11.7\targets\x86_64-linux\lib\stubsで実行- ただ相変わらず同じエラーが出る
- もう一回ビルドをやってみる
mkdir buildをした段階ではもちろんフォルダは空- 次のコードを行うと
buildフォルダの中にファイルが生成cmake -D PKG_GPU=yes -D GPU_API=cuda -D GPU_ARCH=sm_61 -D PKG_GRANULAR=yes -D PKG_KOKKOS=yes -D Kokkos_ARCH_SKX=yes -D Kokkos_ENABLE_OPENMP=yes -D BUILD_OMP=yes -D Kokkos_ARCH_PASCAL61=yes -D Kokkos_ENABLE_CUDA=yes ../cmake - 次のコードを行うと
CMakeCXXCompiler.cmake内の最初の行がset(CMAKE_CXX_COMPILER "/home/masa/lammps-stable_23Jun2022/lib/kokkos/bin/nvcc_wrapper")に置き換わるcmake -D CMAKE_CXX_COMPILER=${HOME}/lammps-stable_23Jun2022/lib/kokkos/bin/nvcc_wrapper ../cmake makeを走らせる(走らせるときには./lmp -in in.foobarとしないとダメ)- やっぱりパッケージが全部入っていない…
..cmakeを常に書き込みしていた…- 正しい手順は
mkdir build cd build cmake ../cmake # ベースとなる構成ファイルをcmakeから取ってくる cmake -D PKG_GPU=yes -D GPU_API=cuda -D GPU_ARCH=sm_61 -D PKG_GRANULAR=yes -D PKG_KOKKOS=yes -D Kokkos_ARCH_SKX=yes -D Kokkos_ENABLE_OPENMP=yes -D BUILD_OMP=yes -D Kokkos_ARCH_PASCAL61=yes -D Kokkos_ENABLE_CUDA=yes -D CMAKE_CXX_COMPILER=${HOME}/lammps-stable_23Jun2022/lib/kokkos/bin/nvcc_wrapper . # 最後は.(ドット)のみ! - 結果は変わらない
LD_LIBRARY_PATHにusr\local\cuda-11.7\targets\x86_64-linux\lib\stubsのパスを追加してもダメだった- ちなみに
PATH=$PATH:/usr/local/cuda-11.7/binの中で$PATHが付いているのは、既存の環境変数を上書きしないようにするため(環境変数は:で複数設定できる) - 環境変数に追加してもだめ、シンボリックリンクを複数作成してもダメ
- ちなみに
- ビルドオプションで
-D CMAKE_CXX_COMPILER=${HOME}/lammps-stable_23Jun2022/lib/kokkos/bin/nvcc_wrapperの部分を外してみた。Illegal instructionの出力が出た- そもそもなぜこのオプションを追加したのか記録が残っていない…
ldd ./lmpで見るとlibcuda.so.1の問題は解決している- あれそうしたら、nvcc_wrapperの部分が問題なのでは??
- もう一度nvccも含めてビルドしなおしたら
Illegal instructionのエラーが出るようになった
- 明日に向けての可能性の整理
- CUDAのインストールがおかしい
- KOKKOSのオプションの設定がおかしい
- cmakeでデバッグオプションみたいなものってつけれれないの?
- 回った…!
- 正解はKOKKOSのオプションでした…
- 今回使っているCPUはIntel Core i9-9900Kで世代としてはCoffee Lakeに相当
- KOKKOSのオプションでは以下のように書かれていて、Coffee Lake世代はSkylake世代よりも後だからArch-IDをSKXに設定していた
Arch-ID | Description BDW | Intel Broadwell Xeon E-class CPU (AVX 2 + transactional mem) SKX | Intel Sky Lake Xeon E-class HPC CPU (AVX512 + transactional mem) - ただIntelのサイトにもあるように9900KはAVX2までしか対応をしていない。そのためArch-IDはBDWに設定するのが正解だった。
- 正解はKOKKOSのオプションでした…
- マジで長かった…
CUDAのドライバエラーに関する解決策
- 今度は別のエラー
- GPUを使ったサンプルコードを回す際に
mpirun -np 1 ./lmp -sf gpu -pk gpu 0 -in ../code/sample2.inを入力 -
Cuda driver error 34 in call at file '/home/ms/lammps-stable_23Jun2022/lib/gpu/geryon/nvd_device.h' in line 326.というエラーが出る。
- このエラーコードはNvidiaのサイトによると
cudaErrorStubLibraryに該当するものらしい - 内容としてはdebug用に使うはずのスタブライブラリを使用しているため、発生するもの
- このサイトが参考になりそう
- おそらくパスをしっかりと設定していないことが原因か?
LD_LIBRARY_PATHを通しても同様のエラーが出る- どうやら
libcuda.soは二つの場所にインストールされるらしいが、今回の場合はstub内にしかインストールされていない
- このエラーコードはNvidiaのサイトによると
- cudaのインストール方法が間違っている?
- インストール方法をNVidiaのこのサイトの方法に変えた
- CUDA 11.7のホスト用ドライバのバージョンを満たしていなかった…(参考サイト)
- アップデートした
- このサイトを参考に
./deviceQueryを動かす- devicequeryのパスは
/usr/local/cuda-11.7/extras/demo_suite
- devicequeryのパスは
- 動いた!
- しかし相変わらず
libcuda.soは一つしかない… - lammpsのコードも動くか…?
- 動いた!
KOKKOS関連のエラーに関する解決策
mpirun -np 1 ./lmp -k on g 1 -sf kk -in ../code/sample2.inのコードを実行したら次の警告のエラーが出た- ```bash
- WARNING: When using a single thread, the Kokkos Serial backend (i.e. Makefile.kokkos_mpi_only) gives better performance than the OpenMP backend (src/KOKKOS/kokkos.cpp:217)
- Kokkos::OpenMP::initialize WARNING: OMP_PROC_BIND environment variable not set
- ERROR: Invalid atom_style command (src/atom_vec.cpp:141) ```
- 1の警告について
- 未調査
- 2の警告について
- 環境変数に
export OMP_PROC_BIND="TRUE"を追加することによって解決した - しかし何をしているのかは良く分かっていない
- 環境変数に
- 3のエラーについて
- atom_styleはKOKKOSの中でもsphereは適用可能なはず
atom_style sphere 0をatom_style sphereにしたら治った
- 上記のエラーが解決後、次のエラーが出た
ERROR: Cannot yet use fix pour with the KOKKOS package (src/GRANULAR/fix_pour.cpp:59)pourコマンドは使えませんよという意味- 代用できるコマンドはいくつかありそうだが、それがKOKKOSに対応しているかどうかをどのように調べればよいのか…
- おそらく
/src/KOKKOS/の中にあるものから一つずつそれっぽいのを探していくしかない… create_atomsコマンドとsetコマンドで乗り切った
- おそらく
- 次のエラー
-
ERROR: Must use half neighbor list with gran/hooke/history/kk (src/KOKKOS/pair_gran_hooke_history_kokkos.cpp:92) - 半分の近傍リストを使えという意味だが、neighbor listとソースコードがよく読み切れていない…
- Lammpsのこのページが使えそう
- 結果として
mpirun -np 1 ../build/lmp -k on g 1 -sf kk -pk kokkos neigh half comm no -in sample2.inのneigh half comm noを追加することによってエラーはでなくなったが、どうやら結果がおかしい。 - 具体的にはこのコマンドを入れると、粒子系全体の周期的な振動が収束しなくなる。
- HWiNFOでGPUの
Reliability VoltageがずっとYesになっている。 - というか
mpirun -n 16にしても全くコアが使用されていない。
-
20220819段階での状況の整理
- CPUのみによる解析は行える(シングルスレッドによる解析)
-
CPU+GPUによる解析は行えるが結果の妥当性の確認が必要
- 今後の方向性
- GPUを使用した更なる高速化
- ` ERROR: Must use half neighbor list~`を無くす
- KOKKOSとLammpsの近傍リスト構造の勉強
mpirunに関する勉強- (上記に付随して)マルチスレッド、タスク、プロセッサーに関する勉強
- ` ERROR: Must use half neighbor list~`を無くす
- CPUのみの解析結果を出す
- 壁面作用応力の抽出
- 粒子位置と作用応力のテキストファイルの抽出
- 可視化手法に関する勉強
- GPUを使用した更なる高速化
20220819の作業メモ
- Neighbor Listsについて
- LAMMPSでは力の効率的な計算のために、ベルレリスト(Verlet List)を用いている。
- ベルレリストとは、分子動力学で使用されるデータ構造である。
- 当該粒子からカットオフ半径(厳密にはカットオフ半径にスキン距離を足した距離)内に存在する粒子のインデックスを作成し、カットオフ半径よりも遠い粒子の相互作用は無視するというもの
- 今回使用している相互作用モデル(
gran/hooke/history)では2粒子の相対距離が2粒子の半径の和を下回った時に作用する力であるため、カットオフ半径はそれほど大きくする必要はない(?)- カットオフ半径は粒子半径+スキン距離でよいかもしれない
- ただタイムステップが長い場合には粒子の移動量が大きくなるため、安定的な解析をするためにはカットオフ半径を大きくする必要があるかもしれない
- 今回使用している相互作用モデル(
- タイムステップ毎に近傍粒子のインデックスを更新するのは計算量を増大させるため、各粒子が等速直線運動をしたと仮定して、その移動距離がスキン距離の半分を超えた段階でリストを更新する
- 実際には粒子はどこかに衝突するので、この予想移動距離は実際の値とは異なる。(ただ予想移動距離が最大値とは限らないのでは…(質量のより重く速度の大きい粒子が真後ろから衝突して運動量が増加するなど)→インデックスを更新したら実際には全部の粒子が外に出てしまっていたなんてこともありうる?)
- スキン距離の更新はor条件(どれか一つの粒子の予想移動距離がSKIN距離の半分を上回ったら更新)
For that atoms are spatially binned and then reordered so that atoms in the same bin are adjacent in the vector.という記述がLAMMPS上のドキュメントにあるがこのベクトルというのはメモリ配列のことを指しているのだろうか?- この並べ替え作業については
atom_modifyコマンドで有効化、無効化できる。- 無効化する必要性は?
atom_modifyドキュメントの中には書かれていないけど、As a general rule, sorting is typically more effective at speeding up simulations of liquids as opposed to solids.と書かれているから粒子の変位量が大きい流体で効果があるのかもしれない
- この並べ替え作業については
- おそらくLAMMPSの
Half Neighbor Listは異なる意味で使われている- Processor≒Sub-domain>binの順?
- ProcessorはMPIから定義されるもので、sub-domainはprocessorが持つ解析領域的なもの
- sub-domainは必ずしも全体の解析領域(entire domain)と平行である必要はない
- binは解析用の格子みたいなもの?
- 全ての粒子タイプのペアのカットオフ半径の最大値の半分が格子サイズとなる。
- ProcessorはMPIから定義されるもので、sub-domainはprocessorが持つ解析領域的なもの
- この図の中には本来は粒子が書かれていたのでは?
- 全ての相互作用の数は $\ _nC_2$ なので近傍リストは総当たりで計算した際の半分で良い
- だから「half」という文字が含まれる
- 粒子の範囲が含まれるような最小の格子の選択がステンシルに相当する
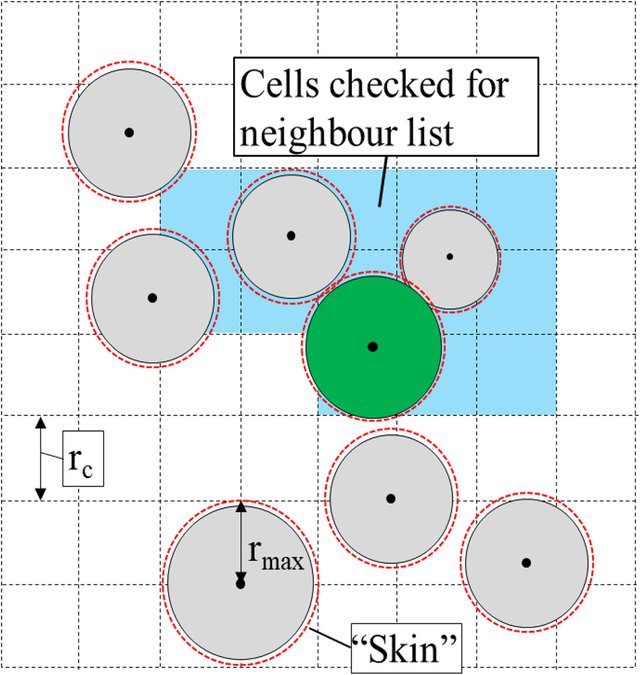
- Processor≒Sub-domain>binの順?
- 以上のことを考えると上記のエラーメッセージは
half neighbor listを使用していないことが問題となっている?
TODO
- 既往文献をあたる(国内でDEMをやられている先生はかなりいる、筑波大松島先生、土研大坪先生)
- マイクロパラメータ(粒子間剛性や粘性、使用している弾塑性モデル)がどのように設定されているのか
- 杭の挿入の際にどのような境界条件を変化させているのか
- 粉体工学のミルの設計に関する研究などが約に立つかもしれない
- 適切な粒子数の設定
- DEMでは粒子数が多くなると解析に必要な時間が増える(解析時間と詳細度のトレードオフ)
- GPGPU(要はグラボ)を導入すると爆発的に速くなるらしいけど、詳細は不明。お金はあるので効果があるようだったら早急に購入したい。
- 初期密度や間隙比をどのように調整するのか
- 粒子に適切な重さを与えることができるのか
- 境界面に作用する応力をデータとして吐き出すことができるのか
- .profileと.bashrcの違い
- CUDAプログラミングの基礎
- MPIの基礎